



Índice de este artículo
- 1 Tipos de priones
- 2 Función de la proteína priónica normal
- 3 ¿Cómo se multiplican o transmiten las proteínas anómalas?
- 4 ¿Fisiopatología de las enfermedades neurodegenerativas?
- 5 Enfermedades neurodegenerativas originadas por priones en el humano
- 6 Enfermedades neurodegenerativas originadas por priones en animales
- 7 Referencias
Gracias al Dr. Stanley Prusiner hoy día se conoce el término “prión” que simboliza la expresión Proteinaceous Infectious Particles, que se traduce como “partícula infecciosa proteinácea”. Los priones (PrP sc) se podrían definir como una proteína anómala que posee una composición idéntica a una proteína neuronal funcional de origen sináptico, que por alguna razón puede evolucionar en algunos individuos, cambiando su configuración espacial, es decir, la manera como esta se pliega entre sí, originando una estructura tridimensional distinta.
Este cambio espacial produce la pérdida de sus funciones, así como también modifica su comportamiento y solubilidad en el tejido nervioso, desencadenando cambios histopatológicos importantes que le dan apariencia de esponja al tejido cerebral, originando la enfermedad neurodegenerativa denominada encefalopatía.
Además, esta proteína posee capacidad para transmitirse de un individuo a otro. Por todas las características antes mencionadas, las enfermedades ocasionadas por priones fueron denominadas encefalopatías espongiformes transmisibles.
Su transmisibilidad generó confusión acerca del agente causal de estas patologías por priones, pues se llegó a pensar que era ocasionada por un virus.
Sin embargo, con posterioridad se supo que se trataba de una proteína que no contiene ácido nucléico, solo es una proteína formada por 253 aminoácidos, cuya información genética de la proteína normal está contenida en el brazo corto del cromosoma 20 en los humanos.
Cabe destacar que, las enfermedades producidas por priones (priónicas) no tienen cura e inevitablemente provocan la muerte, y afectan tanto al humano como a los animales.
Tipos de priones
Se distinguen dos tipos de priones: (PrP c) y (Prp sc).
A las proteínas priónicas normales se le identifica con las siglas (PrP c) y a las proteínas priónicas anómalas se conocen como (PrP sc).
Función de la proteína priónica normal
Aunque es vago este conocimiento, algunos investigadores han revelado que esta proteína en su configuración normal PrP c cumple ciertas funciones en el organismo.
En este sentido, se cree que participan en la reparación del tejido mielínico del sistema nervioso periférico, ya que se ha visto que su ausencia provoca la pérdida de la mielina de las células neuronales. Este proceso se conoce como desmielinización celular.
En experimentos en ratones han demostrado que la desmielinización provoca neuropatía periférica en los animales.
Por su parte, en el desarrollo embrionario parece tener una participación importante en la neuritogénesis (formación de las dendritas y los axones).
Otra función que se le atribuye a la proteína priónica normal (PrP c), es la de neuroprotector, garantizando que haya una irrigación sanguínea adecuada en el cerebro.
Se ha visto que el aumento de la expresión de esta proteína de forma inducida estimula la regeneración en los procesos de isquemia cerebral aguda.
Por otra parte, investigaciones científicas han relacionado a esta proteína como posible coadyuvante en el buen funcionamiento de la memoria, especialmente en la de largo plazo.
Finalmente, debido a que estas proteínas se han encontrado en las membranas de muchas células en el organismo, incluyendo las células madres hematopoyéticas, se cree que desempeñan un papel en la renovación celular.
¿Cómo se multiplican o transmiten las proteínas anómalas?
Existen tejidos en los que estas proteínas anómalas pudieran estar presentes, tanto en humanos como en los animales.
Los principales tejidos son: el cerebro y la médula espinal, sin embargo el contacto con otros tejidos y órganos también podrían resultar infecciosos, como los ojos, las amígdalas y los intestinos.
Por este motivo las personas que manipulan este tipo de tejidos en autopsias, mataderos, etc, deben tomar sus previsiones, para evitar ser infectados, ya que las vías de transmisión son muy variadas y de gran alcance, ya que puede darse entre individuos de una misma especie y de especies diferentes.
En la literatura se han descrito transmisión de priones por la vía oral (alimentos contaminados), parenteral (transfusiones), por absorción transdérmica e intralingual (contacto directo con piel y mucosa) y recientemente según experimentos en ratones la transmisión vía inhalatoria por aerosoles también es posible.
Esto debe llamar la atención para tomar medidas de bioseguridad con mayor cautela, especialmente en los laboratorios donde se manipulan priones o material infectado, más aun cuando se sabe que la proteína anómala es resistente a las proteasas, al calor de la autoclave, a las radicaciones, al formol, al etanol y a otros procesos de esterilización.
Por otra parte, lamentablemente no siempre la infección es exógena, pues en otros casos la enfermedad priónica es de origen espontánea o hereditaria, suscitada por la mutación del gen que contiene la información genética de la (PrP c) y en donde la conversión de la (PrP c) a (PrP sc) se da de forma endógena.
¿Fisiopatología de las enfermedades neurodegenerativas?
La presencia de la proteína anómala en el tejido cerebral adquirida bien sea por transmisión o por mutación, produce una reacción en cadena.
Primero, las proteínas anómalas colonizan el cerebro y luego reclutan a las proteínas priónicas normales (Prp c), convirtiéndolas en anómalas (Prp sc) al inducir un plegamiento anormal, luego estas a su vez repiten el proceso frente a otras (Prp c).
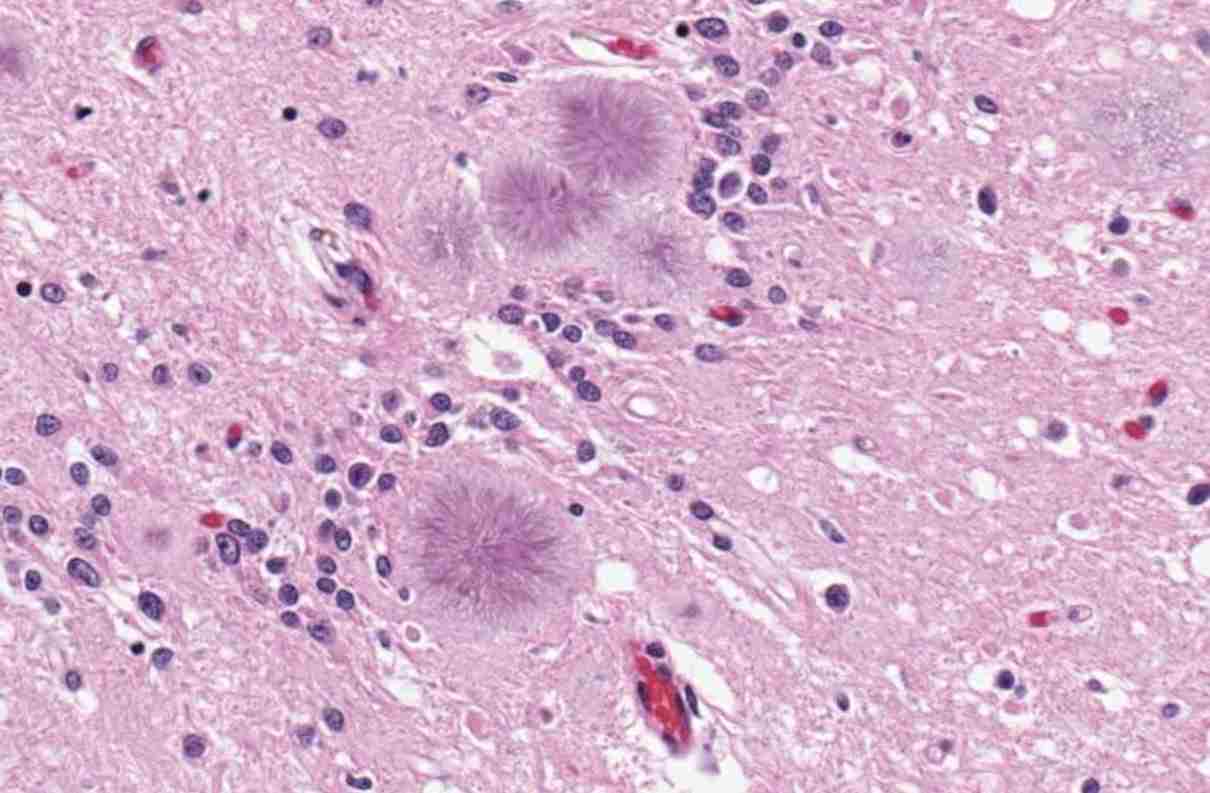
Posteriormente, como las proteínas anómalas son más rígidas y de menor solubilidad que las normales porque poseen un aumento de las láminas β, estas van formando agregados amiloides (amiloidosis) característicos en el tejido cerebral, causando lentamente alteraciones progresivas e irreversibles, hasta provocar la muerte.
En todo el proceso la proteína no estimula al sistema inmunológico porque este la reconoce como propia, por tanto, el organismo es incapaz de defenderse de su presencia.
El aspecto espongiforme que adquiere el tejido cerebral es debido a la formación de vacuolas en las neuronas y a la proliferación marcada de astrocitos (gliosis) que le da esa característica.
Después de un período de incubación largo de 2 a 10 años, comienzan las manifestaciones clínicas, caracterizadas por cambios neurológicos importantes.
El paciente puede presentar falta de coordinación de los movimientos (ataxia), inestabilidad, deficiencia motriz, cambios bruscos de comportamiento, demencia, pérdida de la memoria, insomnio, entre otras. Las manifestaciones clínicas se van agravando conforme pasa el tiempo.
El proceso de estas enfermedades se puede decir que es dinámico, y se resume en las siguientes etapas:
- Ingreso o formación de la proteína anómala
- Replicación
- Neuroinvasión
- Neurodegeneración
Enfermedades neurodegenerativas originadas por priones en el humano
Las patologías neurodegenerativas también se les conoce como enfermedades priónicas, haciendo alusión al origen del problema.
Entre enfermedades originadas por priones más estudiadas se encuentran:
Síndrome de Creutzfeldt-Jakob
Es la más frecuente en los humanos. El paciente presenta disfunción del sistema nervioso central, espasmos musculares, demencia, incoordinación. Existen diversas variantes:
Síndrome de Creutzfeldt-Jakob esporádica (s CJD): Es ocasionada por la mutación del gen de forma aleatoria.
Síndrome de Creutzfeldt-Jakob iatrogénica (iCJD): Ocurre por infección con material contaminado, bien sea durante un trasplantes de duramadre o de cornea. También por recibir tratamiento con hormona de crecimiento o por contacto con electrodos de neurocirugía contaminados.
Síndrome de Creutzfeldt-Jakob familiar (fCDJ): Es de carácter hereditario autosómica dominante, por tanto aparece debido a la genética del individuo.
Síndrome de Creutzfeldt-Jakob variante (vCJD): Ocurre por consumo de alimentos contaminados (transmisión oral). Principalmente carne de bovinos con encefalopatía espongiforme bovina.
Kuru
Se asocia al canibalismo (consumo de carne y cerebro humano) en la región de Papúa Nueva Guinea.
La característica más notable es el temblor incontrolado, además de dolores musculares, incoordinación, dificultad para tragar, dolor de cabeza, entre otras. La muerte se presenta a los 2 años aproximadamente.
Enfermedad de Gerstmann-Sträussler-Scheinker
Esta enfermedad originada por priones es de carácter hereditario autosómico dominante.
El paciente presenta movimientos violentos involuntarios, con actitud agresiva, inestabilidad, demencia, pérdida de la audición, dificultad para pronunciar las palabras, disminución de la respuesta refleja (hiporreflexia), entre otros. La supervivencia alcanza los 5 años.
Prionopatía con sensibilidad variable a la proteasa
Similar a la enfermedad de Gerstmann-Sträussler- Scheinker. La proteína priónica que causa esta enfermedad posee mayor sensibilidad a las proteasas de allí su nombre, eso la diferencia del resto de las enfermedades neurodegenerativas.
Generalmente aparece a partir de los 70 años y el paciente no sobrevive más de 2 años después de iniciado los síntomas.
Alzheimer
Esta enfermedad originada por priones es bastante frecuente. Se caracteriza por la pérdida progresiva de habilidades cognitivas y de la memoria. La esperanza de vida después de su diagnóstico oscila entre 4 a 20 años con un promedio de 8 años.
Como todas estas enfermedades no tiene cura, pero existen tratamientos que enlentecen el progreso de la enfermedad, especialmente si se hace un diagnóstico temprano, mejorando así la calidad de vida del paciente.
La demencia frontotemporal
El grupo de enfermedades que encajan en esta categoría se denominan así porque afecta principalmente el área de los lóbulos frontal y temporal.
Suele afectar a individuos entre 40 a 60 años. Entre algunas de las afecciones se pueden mencionar: problemas con el habla (afasia progresiva), dificultad para comprender el lenguaje (demencia semántica), disminución de la movilidad (degeneración corticobasal), enfermedad de Pick (anormalidad en la proteína tau).
Las enfermedades de Huntington y Parkinson
El Huntington es una enfermedad hereditaria que cursa con degeneración de las neuronas. Afecta tanto el aprendizaje, el movimiento y la salud mental. Es frecuente en personas jóvenes (30 a 40 años de edad e incluso antes).
Por su parte, el Parkinson es una enfermedad que afecta principalmente la parte motora por lo que la característica más notable es el temblor involuntario. También se describe una disminución de la fuerza muscular y doblez del tronco (espalda jorobada).
Esclerosis lateral amiotrófica
Se conoce también como enfermedad de Lou Gehrig. Esta enfermedad produce un debilitamiento intenso a nivel de los músculos, lo que impide al paciente mover extremidades y el cuerpo en general. En la etapa terminal los músculos del tórax se debilitan, dificultando la respiración severamente.
El insomnio familiar fatal
Enfermedad muy rara y poco frecuente que tiene dos variantes: una hereditaria y otra por mutación espontánea.
Como su nombre lo indica el paciente presenta imposibilidad para dormir (insomnio permanente), ocasionado por la desaparición o disfunción del sistema reticular descendente, que se encarga de inhibir al sistema reticular ascendente encargado de la vigilia.
Esta situación genera un gran deterioro que incluye: pérdida de la memoria, problemas motores (ataxia), espasmos musculares, pérdida de peso, depresión, alucinaciones, sudoración excesiva, taquicardia, entre otros síntomas. El paciente no dura más de 6 años.
Enfermedades neurodegenerativas originadas por priones en animales
Las enfermedades neurodegenerativas originadas por priones, también afectan a diversos animales, la principal causa de transmisión es la oral. Se producen cambios psíquicos y motores en el animal afectado. Entre ellas se encuentran:
- La encefalopatía espongiforme bovina.
- Scrapie de ovejas y cabras.
- Encefalopatía de los visones.
- Encefalopatía espongiforme de mulas y ciervos.
- Enfermedad del Desgaste Crónico.
- Encefalopatía Espongiforme Felina.
Referencias
- Haybaeck J, Heikenwalder M, Klevenz B, Schwarz P, Margalith I, Bridel C, Mertz K, et al. Los aerosoles transmiten priones a ratones inmunocompetentes e inmunodeficientes. PLoS Pathog . 2011; 7 (1): e1001257. Available in:ncbi.nlm
- Toro González Gabriel, Sierra Zuleta Uriel Esteban, Gómez Grosso Luis Alberto. Acta Neurol Colomb. [Internet]. Enero de 2015 [consultado el 7 de septiembre de 2020]; 31 (1): 101-112. Disponible en: scielo
- Priones y enfermedades espongiformes transmisibles. Salud pública de México; 2001, 43 (3): 257-258. Disponible en: scielosp.org/pdf
- Colby DW, Prusiner SB. De novo generation of prion strains. Nat Rev Microbiol, 2011; 9 (11): 771-7. Available in:ncbi.nlm.nih.gov/
- Farías G, Garces H, Larenas J, Ramirez A, Lecocq C. Enfermedades Producidas Por Priones en los Animales. Avances en Ciencias Veterinarias, 2011; 26 (1):1-10 Disponible en: repositorio.uchile.
Otros temas relacionados
- Qué es el albinismo, tipos y herencia asociada


- Proteínas, qué son, funciones y estructura. Clasificación


- Composición química de los seres vivos: biomoléculas…


- ¿Qué es un gen? ¿Para qué sirve? Estructura


- Mutaciones genéticas, qué son, tipos, causas y ejemplos

- Ácido ribonucleico, qué es, estructura, funciones y tipos


Deja una respuesta